Рубрика МКБ-10: Q87.4
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q80-Q89 Другие врожденные аномалии пороки развития / Q87 Другие уточненные синдромы врожденных аномалий пороков развития, затрагивающих несколько систем
Определение и общие сведения[править]
Синдром Марфана
Синдром Марфана — редкое системно-наследственное заболевание, при котором наблюдают аномалии развития костной, сердечнососудистой систем и органа зрения.
В 1896 г. в Париже профессор педиатрии Антонин-Бернард Марфан описал пятилетнюю девочку с диспропорционально длинными конечностями и пальцами (pattes d’araignee), фиброзными контрактурами пальцев и коленных суставов, высоким ростом и узким удлиненным черепом (долихоцефалия). Патологии сердца, глазного яблока и интеллекта не отмечалось. Сорока годами позже в наблюдениях профессора Марфана уже насчитывалось более 150 сходных случаев этой патологии с долихостеномелией (термин, введенный Марфаном для обозначения необычно длинных конечностей). Со временем стало известно необычное поражение других тканей — подвывих хрусталика и дисфункция митрального клапана сердца. В 1943 г. было описано серьезное поражение аорты, и к 1972 г. стала окончательно ясна роль этого поражения для жизненного прогноза пациентов (Murdoch et al., 1972). В своей монографии 1956 г. Маккьюсик предположил, что основной дефект соединительный ткани является общим для поражения как интимы аорты, так и глазного яблока. Однако только через 36 лет было показано, что мутация гена фибриллина 1 (FBN1), кодирующего гликопротеин фибриллин 1-го типа — основной компонент экстрацеллюлярного матрикса микрофибрилл, является первичным патологическим звеном для классического синдрома Марфана (Dietz et al., 1991). Очевидные успехи применения β-блокаторов для предотвращения возникновения аневризмы аорты и своевременной операции по замещению пораженного участка аорты протезом позволили увеличить среднюю продолжительность жизни этих пациентов с 43 лет (70-е годы ХХ в.) до нормальных 73 лет (90-е годы ХХ в.).
Распространенность оценивается в 1/5000, оба пола в равной степени подвержены заболеванию. Наследуется аутосомно-доминантно.
Этиология и патогенез[править]
В подавляющем большинстве случаев синдром Марфана вызван мутациями гена FBN1 (15q21), который кодирует белок фибриллин-1, необходимый для формирования соединительных тканей. Были идентифицированы также пограничные формы синдрома Марфана, которые являются вторичными по отношению к мутациям в гене TGFBR2, расположенный на хромосоме 3, который кодирует рецептор TGF-бета.
Клинические проявления[править]
Особенности внешнего вида: арахнодактилия (длинные и тонкие фаланги пальцев рук и ног), удлинение самих конечностей, слабость связочного аппарата, куриная грудь, искривление позвоночника, долихоцефалический череп. Бывают пороки сердца, аневризма аорты, тромбозы вен. В некоторых случаях выявляют тяжелые эндокринные нарушения и нервно-психические расстройства. Со стороны глаз наблюдают вывихи хрусталика в переднюю камеру или стекловидное тело, в результате может развиться вторичная глаукома.
При синдроме Марфана при подвывихе хрусталик смещается вверх, бывают также микрофакия, колобома хрусталика.
Отмечают иридодонез, миоз, могут быть мегалокорнеа или микрокорнеа, высокая степень близорукости, атрофия зрительного нерва. Встречаются случаи пигментной дегенерации сетчатки.
- Читайте также:
Синдром Марфана: Диагностика[править]
Условия диагностики синдрома Марфана:
• при отсутствии пораженного родственника (мать, отец, сибс, ребенок) и негативных ДНК тестов (FBN1) должен присутствовать главный признак в двух различных системах и один малый признак из третьей системы;
• при выявлении мутации гена FBN1 достаточно одного главного критерия любой системы и вовлечения еще одной системы.
Физикальное обследование
Диагностические признаки следующие.
• Скелет:
— главные признаки — килевидная деформация грудной клетки, воронкообразная деформация грудной клетки III-IV степени, уменьшение соотношения верхнего/нижнего сегмента тела или увеличение соотношения размаха конечности роста более чем на 1,05, симптомы запястья и I пальца кисти, сколиоз (угол более 20°) или спондилолистез, ограничение разгибания в локтевом суставе (менее 170°), медиальное смещение медиальной лодыжки, плоскостопие, протрузия вертлужной впадины любой степени (рентгенологическое исследование);
— малые признаки — воронкообразное вдавление грудины, гипермобильность суставов, высокое нёбо с неправильным ростом зубов, черепно-лицевые аномалии (долихоцефалия, гипоплазия скуловых дуг, энофтальм, ретрогнатия, антимонголоидный разрез глаз). Скелетная система считается вовлеченной, если присутствуют два признака из главных или один из главных и два из малых признаков.
• Глазное яблоко:
 Читайте также:
Читайте также:
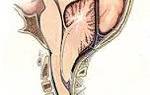
— главный признак — эктопия хрусталика;
— малый признак — уплощенная роговица (кератометрия), увеличение оси глазного яблока (УЗИ), гипоплазия радужки или цилиарной мышцы, приводящая к снижению миоза. Глазная система считается вовлеченной, если присутствуют два малых признака.
Читайте также: Преподавание математики детям с синдромом дауна
• Сердечно-сосудистая система:
— главные признаки — расширение восходящей аорты с наличием или отсуствием аортальной регургитации и вовлечением синуса Вальсальвы, расслоение стенки восходящей аорты;
— малые признаки — пролапс митрального клапана, расширение легочной артерии при отсутствии какой-либо причины клапанного или периферического стеноза в возрасте после 40 лет; кальцификация митрального кольца после 40 лет, расширение или расслоение нисходящего отдела грудной или брюшной аорты в возрасте старше 50 лет. Система считается вовлеченной при наличии одного главного или одного малого признака.
• Легочная система:
— главные признаки — отсутствуют;
— малые признаки — спонтанный пневмоторакс, апикальные псевдокисты легкого (рентгенография).
- Читайте также:
Легочная система считается вовлеченной при наличии одного малого признака.
• Кожа и наружные покровы:
— главные признаки — отсутствуют;
— малые признаки — атрофичные стрии, не связанные с колебаниями массы тела, беременностью или физическими растяжениями, рецидивирующие грыжи любой локализации.
Система считается вовлеченной при наличии одного главного или одного малого критерия.
• Твердая мозговая оболочка:
— главный признак — пояснично-крестцовое расширение (эктазия) эпидурального пространства (рентгеновская или компьютерная томография спинного мозга).
Инструментальные исследования
Рентгенологическое исследование и КТ.
- Читайте также:
Показания к консультации других специалистов
Все больные с синдромом Марфана должны наблюдаться у кардиолога, офтальмолога и ортопеда.
Наиболее важные аспекты этой патологии для неонатологов и хирургов:
• затруднение интубации из-за подвижности височнонижнечелюстного сустава и шейного отдела позвоночника;
• опасность внезапного повышения или снижения АД во время операции;
• осторожное применение мышечных релаксантов при миопатическом синдроме (возможен парадоксальный или пролангированный эффект);
• возможность летальной желудочковой аритмии и бактериального эндокардита в послеоперационном периоде при пролапсе клапанов сердца с регургитацией;
• разрыв аневризмы аорты;
• повышенный риск спонтанного пневмоторакса (5%);
• повышенная частота пневмоний и хронических эмфиземоподобных изменений;
• снижение жизненной емкости легких, увеличивающее риск анестезиологических осложнений.
Нестабильность шейного отдела позвоночника изредка создает проблемы, что необходимо учитывать анестезиологам при проведении эндотрахеальной интубации, но истинная частота встречаемости этой патологии неизвестна.
Дифференциальный диагноз[править]
Дифференциальный диагноз включает MASS-синдром, синдром Шпринтцена-Гольдберга, пролапс митрального клапана, синдром Элерса-Данло и другие заболевания, которые сопровождаются аневризмой аорты, например, синдром Лойса-Дица.
Синдром Марфана: Лечение[править]
Немедикаментозное лечение
У большинства больных деформация позвоночника развивается постепенно в течение детского периода. При наличии кифоза и сколиоза умеренной тяжести показано консервативное лечение.
Медикаментозное лечение
Прием β-адреноблокаторов (пропранолол) в целях профилактики аневризмы аорты.
Хирургическое лечение
Патологическая деформация позвоночника может возникнуть на любом уровне и в любом возрасте. Иногда новорожденные имеют тяжелую деформацию, требующую инструментального
вмешательства. Опасность раннего хирургического лечения заключается в разрушении зон роста позвонков и снижении высоты грудной клетки.
При сколиотической деформации, превышающей 40°, показана хирургическая стабилизация позвоночника.
Профилактика[править]
Прочее[править]
Синдром Лойса-Дитца
Синонимы: синдром аневризмы аорты в связи с аномалиями TGF-бета-рецепторов
Синдром Лойса-Дитца это редкое генетическое заболевание соединительной ткани, которое характеризуется широким спектром краниофациальных, скелетных и сосудистых проявлений.
Синдром Лойса-Дитца имеет четыре генетических подтипа, наследуется аутосомно-доминантно.
Источники (ссылки)[править]
Неонатология. Национальное руководство. Краткое издание [Электронный ресурс] / Под ред. Н.Н. Володина — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970424438.html
Офтальмология [Электронный ресурс] : учебник / Тахчиди Х.П., Ярцева Н.С., Гаврилова Н.А., Деев Л.А. — М. : ГЭОТАР-Медиа, 2011. — https://www.rosmedlib.ru/book/ISBN9785970418208.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
СиндÑом ÐаÑÑана (или аÑÐ°Ñ Ð½Ð¾Ð´Ð°ÐºÑилиÑ) â ÑÑо наÑледÑÑвенное заболевание, Ñ Ð°ÑакÑеÑизÑÑÑееÑÑ Ð½ÐµÐ´Ð¾ÑÑаÑоÑноÑÑÑÑ ÑоединиÑелÑной Ñкани. ÐÐ¾Ð»ÐµÐ·Ð½Ñ Ð¿ÑÐ¸Ð²Ð¾Ð´Ð¸Ñ Ðº паÑологиÑеÑким изменениÑм ÑеÑдеÑно-ÑоÑÑдиÑÑой, неÑвной, опоÑно-двигаÑелÑной и дÑÑÐ³Ð¸Ñ ÑиÑÑем и оÑганов.
ÐÑиÑÐ¸Ð½Ñ Ð²Ð¾Ð·Ð½Ð¸ÐºÐ½Ð¾Ð²ÐµÐ½Ð¸Ñ ÑиндÑома ÐаÑÑана

ÐÑÐ°Ñ Ð½Ð¾Ð´Ð°ÐºÑÐ¸Ð»Ð¸Ñ Ð½Ð°ÑледÑеÑÑÑ Ð¿Ð¾ аÑÑоÑомно-доминанÑÐ½Ð¾Ð¼Ñ ÑипÑ, поÑÑÐ¾Ð¼Ñ Ð²ÑÑÑеÑаеÑÑÑ Ð¿Ð¾ÑÑи в одинаковом ÑооÑноÑении, как Ñ Ð¼ÑжÑин, Ñак и Ñ Ð¶ÐµÐ½Ñин. ÐÑо доÑÑаÑоÑно Ñедкое генеÑиÑеÑкое заболевание Ñ ÑаÑÑоÑой Ð²Ð¾Ð·Ð½Ð¸ÐºÐ½Ð¾Ð²ÐµÐ½Ð¸Ñ 1:5000.
ÐногоÑиÑленнÑе иÑÑÐ»ÐµÐ´Ð¾Ð²Ð°Ð½Ð¸Ñ Ð¿Ð¾ÐºÐ°Ð·Ð°Ð»Ð¸, ÑÑо ÑиндÑом ÐаÑÑана обÑÑловлен мÑÑаÑией в 15-й Ñ ÑомоÑоме гена белка ÑибÑиллина, вÑледÑÑвие Ñего наÑÑÑаеÑÑÑ ÑÑÑÑкÑÑÑа и вÑÑабоÑка коллагена.
Ðо ÑÑаÑиÑÑике в 75% ÑлÑÑаев мÑÑиÑовавÑий ген пеÑедаеÑÑÑ Ð´ÐµÑÑм Ð¾Ñ Ð·Ð°Ð±Ð¾Ð»ÐµÐ²ÑÐ¸Ñ ÑодиÑелей, а в оÑÑалÑнÑÑ ÑлÑÑаÑÑ Ðº болезни пÑиводÑÑ ÑпонÑанно возникÑие в Ð¼Ð¾Ð¼ÐµÐ½Ñ Ð·Ð°ÑаÑÐ¸Ñ Ð³ÐµÐ½ÐµÑиÑеÑкие мÑÑаÑии.
Читайте также: Синдром короткой кишки у ребенка
ÐÑиÑÐ¸Ð½Ñ Ð¿Ð¾Ð´Ð¾Ð±Ð½ÑÑ «Ð¿Ð¾Ð»Ð¾Ð¼Ð¾Ðº» в Ð³ÐµÐ½Ð°Ñ Ð´Ð¾ конÑа Ñак и не вÑÑÑненÑ, но Ñ Ð²ÐµÑоÑÑноÑÑÑÑ Ð² 50% можно ÑÑвеÑждаÑÑ, ÑÑо деÑи, ÑожденнÑе Ñ ÑиндÑомом ÐаÑÑана, пеÑедадÑÑ ÑÑо заболеванием поÑомкам.
ÐÑизнаки ÑиндÑома ÐаÑÑана
Ð ÑамÑм замеÑнÑм ÑимпÑомам болезни оÑноÑÑÑÑÑ Ð²Ð½ÐµÑние Ð¸Ð·Ð¼ÐµÐ½ÐµÐ½Ð¸Ñ Ð¾Ð±Ð»Ð¸ÐºÐ° Ñеловека. Так, заболевÑие лÑди имеÑÑ Ð²ÑÑокий ÑоÑÑ Ð¾ÑноÑиÑелÑно ÑÐ²Ð¾Ð¸Ñ ÑодÑÑвенников. ÐÑÑгой пÑизнак, бÑоÑаÑÑийÑÑ Ð² глаза â ÑÑо аÑÑениÑеÑкое ÑелоÑложение, пÑи коÑоÑом конеÑноÑÑи гоÑаздо длиннее, а Ñакже наблÑдаеÑÑÑ Ð°ÑÐ°Ñ Ð½Ð¾Ð´Ð°ÐºÑилиÑ, или «Ð¿Ð°ÑÑÑи палÑÑÑ». У вÑÐµÑ Ð»Ñдей Ñ ÑÑим заболеванием ÑдлиненнÑй ÑеÑеп, маленÑÐºÐ°Ñ ÑелÑÑÑÑ Ñ Ð½ÐµÐ¿ÑавилÑно ÑаÑÑÑÑими зÑбами и глÑбоко поÑаженнÑе глаза.
ÐбÑÑно пÑизнаки ÑиндÑома ÐаÑÑана ÑазделÑÑÑ Ð½Ð° гÑÑÐ¿Ð¿Ñ Ð² ÑооÑвеÑÑÑвии Ñ Ð¾Ñганами и ÑиÑÑемами, в коÑоÑÑÑ Ð¾Ð½Ð¸ пÑоÑвлÑÑÑÑÑ. ÐапÑимеÑ, вÑÑокий ÑоÑÑ Ð¸ ÑдлиненнÑе конеÑноÑÑи â ÑÑо пÑоÑÐ²Ð»ÐµÐ½Ð¸Ñ Ñо ÑÑоÑÐ¾Ð½Ñ Ð¾Ð¿Ð¾Ñно-двигаÑелÑного аппаÑаÑа. Также к ним оÑноÑÑÑÑÑ Ð¸ÑкÑивление позвоноÑника, деÑоÑмаÑÐ¸Ñ Ð³ÑÑдной клеÑки, мÑгкоÑÑÑ ÑÑÑÑавов и плоÑкоÑÑопие.
СимпÑÐ¾Ð¼Ñ Ñо ÑÑоÑÐ¾Ð½Ñ Ð³Ð»Ð°Ð· â помÑÑнение и вÑÐ²Ð¸Ñ Ñ ÑÑÑÑалика (его ÑмеÑение Ñ ÐµÑÑеÑÑвенного меÑÑоположениÑ), близоÑÑкоÑÑÑ, оÑÑлаивание ÑеÑÑаÑки и повÑÑенное внÑÑÑиглазное давление.
Ðаиболее опаÑнÑми пÑизнаками ÑиндÑома ÐаÑÑана ÑвлÑÑÑÑÑ ÑимпÑомÑ, каÑаÑÑиеÑÑ ÑеÑдеÑно-ÑоÑÑдиÑÑой ÑиÑÑемÑ. Ðаболевание пÑÐ¸Ð²Ð¾Ð´Ð¸Ñ Ðº ÑаÑÑÐ»Ð¾ÐµÐ½Ð¸Ñ ÑÑенок аоÑÑÑ Ð¸ ÑаÑÑиÑÐµÐ½Ð¸Ñ ÐµÐµ коÑнÑ, а ÑÑо ÑÑеваÑо неожиданнÑм ÑазÑÑвом главной аÑÑеÑии ÑеÑдÑа и пÑакÑиÑеÑки неминÑемÑм леÑалÑнÑм иÑÑ Ð¾Ð´Ð¾Ð¼. ÐÑоме Ñого, пÑи болезни ÐаÑÑана иногда наблÑдаеÑÑÑ Ð½ÐµÐ¿Ð»Ð¾Ñное закÑÑÑие клапанов ÑеÑдÑа, ÑÑо пÑÐ¸Ð²Ð¾Ð´Ð¸Ñ Ðº ÑвелиÑÐµÐ½Ð¸Ñ ÐµÐ³Ð¾ ÑазмеÑов, ÑÑмам, одÑÑке и неÑегÑлÑÑÐ½Ð¾Ð¼Ñ ÑеÑдÑебиениÑ.
Ðенее ÑеÑÑезнÑми ÑÑиÑаÑÑÑÑ Ð¿ÑоÑÐ²Ð»ÐµÐ½Ð¸Ñ Ð¿Ð°Ñологии Ñо ÑÑоÑÐ¾Ð½Ñ Ð»ÐµÐ³ÐºÐ¸Ñ , кожи и неÑвной ÑиÑÑемÑ.
ÐиагноÑÑика ÑиндÑома ÐаÑÑана
ÐиагноÑÑиÑоваÑÑ ÑиндÑом ÐаÑÑана можно лиÑÑ Ð¿Ð¾Ñле комплекÑного обÑÐ»ÐµÐ´Ð¾Ð²Ð°Ð½Ð¸Ñ ÑпеÑиалиÑÑами Ñазного пÑоÑилÑ: каÑдиологом, оÑÑопедом, оÑÑалÑмологом и генеÑиком.
ÐÑаÑ, ÑпеÑиализиÑÑÑÑийÑÑ Ð½Ð° наÑледÑÑвеннÑÑ Ð½Ð°ÑÑÑениÑÑ , изÑÑÐ¸Ñ Ð¸ÑÑоÑÐ¸Ñ ÑемÑи Ñ ÑелÑÑ Ð²ÑÑÐ²Ð»ÐµÐ½Ð¸Ñ ÑÐµÑ ÑодÑÑвенников, коÑоÑÑе ÑмеÑли Ð¾Ñ ÑеÑдеÑно-ÑоÑÑдиÑÑÑÑ Ð·Ð°Ð±Ð¾Ð»ÐµÐ²Ð°Ð½Ð¸Ð¹.
ÐаÑдиолог назнаÑÐ¸Ñ ÑенÑген гÑÑдной клеÑки и измеÑение ÑлекÑÑиÑеÑкой акÑивноÑÑи ÑеÑдÑа (ÐÐÐ). Также Ð½ÐµÐ¾Ð±Ñ Ð¾Ð´Ð¸Ð¼Ð° ÑÑ Ð¾ÐºÐ°ÑдиогÑамма, на оÑнове коÑоÑой бÑдÑÑ Ð¿Ð¾Ð»ÑÑÐµÐ½Ñ ÑазмеÑÑ Ð°Ð¾ÑÑÑ Ð¸ пÑовеÑено ÑÑнкÑиониÑование клапанов.
ÐÑаÑ-оÑÑопед опÑеделиÑ, еÑÑÑ Ð»Ð¸ иÑкÑивление гÑÑдной клеÑки и позвоноÑника, плоÑкоÑÑопие и дÑÑгие обÑие пÑоблемÑ.
ÐÑмоÑÑ Ð³Ð»Ð°Ð· оÑÑалÑмологом Ð¿Ð¾Ð·Ð²Ð¾Ð»Ð¸Ñ Ð¾Ð±Ð½Ð°ÑÑжиÑÑ Ð¿Ð°ÑологиÑеÑкие Ð¸Ð·Ð¼ÐµÐ½ÐµÐ½Ð¸Ñ Ñ ÑÑÑÑалика, еÑли ÑаковÑе имеÑÑÑÑ.
ÐÑиÑеÑии диагноÑÑики ÑиндÑома ÐаÑÑана доÑÑаÑоÑно ÑÑÑогие и ÑоÑнÑе. ÐÑо ÑвÑзано Ñ Ñем, ÑÑо ÑÑд оÑобенноÑÑей болезни ÑипиÑен (напÑимеÑ, не вÑегда вÑÑокий и Ñ Ñдой Ñеловек Ñ Ð´Ð»Ð¸Ð½Ð½Ñми Ñонкими палÑÑами болен). ÐÑоме Ñого, ÑÑÑеÑÑвÑÑÑ Ð¸ дÑÑгие Ð·Ð°Ð±Ð¾Ð»ÐµÐ²Ð°Ð½Ð¸Ñ ÑоединиÑелÑной Ñкани, коÑоÑÑе ÑопÑовождаÑÑÑÑ Ð¿Ð¾Ñ Ð¾Ð¶Ð¸Ð¼Ð¸ ÑимпÑомами.
ÐконÑаÑелÑнÑй диагноз завиÑÐ¸Ñ Ð¾Ñ Ð¿Ð¾Ð´ÑвеÑÐ¶Ð´ÐµÐ½Ð¸Ñ Ð¸Ð»Ð¸ опÑовеÑÐ¶ÐµÐ½Ð¸Ñ Ð°ÑÐ°Ñ Ð½Ð¾Ð´Ð°ÐºÑилии в Ñемейном анамнезе. ÐÑли Ñ ÐºÐ¾Ð³Ð¾-Ñо из ÑодÑÑвенников бÑла ÑÑа болезнÑ, доÑÑаÑоÑно подÑвеÑÐ¶Ð´ÐµÐ½Ð¸Ñ Ð² двÑÑ ÑиÑÑÐµÐ¼Ð°Ñ Ð¾Ñганизма, в пÑоÑивном ÑлÑÑае â в ÑÑÐµÑ .
СимпÑÐ¾Ð¼Ñ Ð·Ð°Ð±Ð¾Ð»ÐµÐ²Ð°Ð½Ð¸Ñ Ð½Ðµ пÑоÑвлÑÑÑÑÑ Ð² Ñаннем деÑÑÑве, а обнаÑÑживаÑÑÑÑ Ð»Ð¸ÑÑ Ñ Ð²Ð¾Ð·ÑаÑÑом. СвоевÑÐµÐ¼ÐµÐ½Ð½Ð°Ñ Ð´Ð¸Ð°Ð³Ð½Ð¾ÑÑика ÑиндÑома ÐаÑÑана оÑÐµÐ½Ñ Ð²Ð°Ð¶Ð½Ð°, но даже еÑли диагноз ÑÑавиÑÑÑ Ð³Ð¾Ð´Ð°Ð¼Ð¸, леÑиÑÑ Ð¿Ñизнаки можно еÑе до оконÑаÑелÑного веÑдикÑа вÑаÑей.
ÐеÑение ÑиндÑома ÐаÑÑана
Ðа ÑегоднÑÑний Ð´ÐµÐ½Ñ Ð½Ðµ ÑÑÑеÑÑвÑÐµÑ ÑÑÑекÑивнÑÑ Ð¼ÐµÑодик леÑÐµÐ½Ð¸Ñ ÑиндÑома ÐаÑÑана. ÐÑе меÑопÑиÑÑÐ¸Ñ ÑводÑÑÑÑ Ðº пÑедÑпÑÐµÐ¶Ð´ÐµÐ½Ð¸Ñ Ð¾Ñложнений болезни.

ЧÑÐ¾Ð±Ñ Ð¿ÑедоÑвÑаÑиÑÑ Ð¾Ð¿Ð°ÑнÑе аоÑÑалÑнÑе изменениÑ, назнаÑаеÑÑÑ Ð¿ÑепаÑÐ°Ñ ÐнапÑилин, но его ÑÑÑекÑивноÑÑÑ Ð½Ðµ доказана. Ðногда пÑоводиÑÑÑ Ñ Ð¸ÑÑÑгиÑеÑÐºÐ°Ñ Ð¿Ð»Ð°ÑÑика аоÑÑÑ Ð¸ клапанов ÑеÑдÑа. ÐеÑеменнÑм женÑинам Ñ Ð±Ð¾Ð»ÐµÐ·Ð½ÑÑ ÐаÑÑана и вÑÑаженной паÑологией Ñо ÑÑоÑÐ¾Ð½Ñ ÑеÑдеÑно-ÑоÑÑдиÑÑой ÑиÑÑÐµÐ¼Ñ Ð¿ÑоводÑÑ Ð¿Ð»Ð°Ð½Ð¾Ð²Ð¾Ðµ кеÑаÑево ÑеÑение. ÐÐ»Ñ Ð¿ÑоÑилакÑики ÑÑомбозов и инÑекÑионного ÑндокаÑдиÑа поÑле опеÑаÑивного вмеÑаÑелÑÑÑва назнаÑаÑÑÑÑ Ð°Ð½ÑикоагÑлÑнÑÑ Ð¸ анÑибиоÑики.
ÐÑи ÑилÑном Ñколиозе Ð¿Ð¾ÐºÐ°Ð·Ð°Ð½Ñ ÑизиоÑеÑапевÑиÑеÑкие пÑоÑедÑÑÑ, а Ñакже Ð¼ÐµÑ Ð°Ð½Ð¸ÑеÑкое ÑкÑепление ÑкелеÑа. ХиÑÑÑгиÑеÑÐºÐ°Ñ ÐºÐ¾ÑÑекÑÐ¸Ñ Ð¿ÑоводиÑÑÑ Ð² Ñом ÑлÑÑае, еÑли Ñгол оÑÐºÐ»Ð¾Ð½ÐµÐ½Ð¸Ñ Ð¿Ð¾Ð·Ð²Ð¾Ð½Ð¾Ñника ÑоÑÑавлÑÐµÑ 45° и более.
ÐÑогноз жизни лÑдей Ñ ÑиндÑомом ÐаÑÑана завиÑиÑ, в пеÑвÑÑ Ð¾ÑеÑедÑ, Ð¾Ñ ÑÑжеÑÑи и ÑÑепени изменений, заÑÑагиваÑÑÐ¸Ñ ÑеÑдеÑно-ÑоÑÑдиÑÑÑÑ ÑиÑÑемÑ. ÐÑи ÑазÑÑве легоÑного ÑÑвола и аневÑизме аоÑÑÑ Ð² болÑÑинÑÑве ÑлÑÑаев паÑÐ¸ÐµÐ½Ñ ÑмиÑаеÑ.
ÐеÑение ÑиндÑома ÐаÑÑана подÑазÑÐ¼ÐµÐ²Ð°ÐµÑ Ð¿Ð¾ÑÑоÑннÑй вÑаÑебнÑй конÑÑÐ¾Ð»Ñ Ð¸ ÑегÑлÑÑное диагноÑÑиÑеÑкое обÑледование. ФизиÑеÑÐºÐ°Ñ Ð°ÐºÑивноÑÑÑ Ð±Ð¾Ð»ÑнÑÑ Ð´Ð¾Ð»Ð¶Ð½Ð° бÑÑÑ Ñнижена до ÑÑеднего или низкого ÑÑовнÑ, Ñо еÑÑÑ ÑледÑÐµÑ Ð¸ÑклÑÑиÑÑ ÑпоÑÑивнÑе ÑоÑевнованиÑ, конÑакÑнÑе Ð²Ð¸Ð´Ñ ÑпоÑÑа, подводное плавание и изомеÑÑиÑеÑкие нагÑÑзки. ÐенÑинам деÑоÑодного возÑаÑÑа, имеÑÑим данное заболевание, Ð½ÐµÐ¾Ð±Ñ Ð¾Ð´Ð¸Ð¼Ð¾ обÑзаÑелÑно пÑойÑи медико-генеÑиÑеÑкое конÑÑлÑÑиÑование.